Execute a integração de dados do Seurat
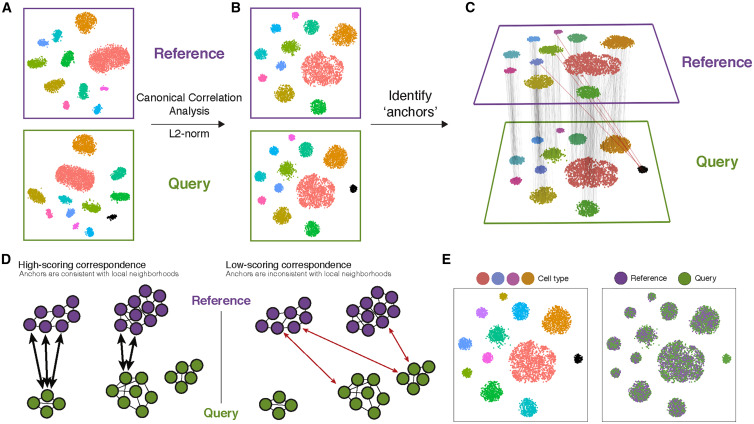
A integração aqui realizada com Seurat consiste na utilização de uma Análise de Correlação Canônica para identificar âncoras entre conjuntos de dados, conforme indicado no original paper:
Dividir conjunto de dados em uma lista de conjuntos de dados
# Dividir (split) o objeto Seurat em uma lista de objetos Seurat menores com base na coluna de metadados "stim"
sc_datasets.list <- SplitObject(sc_datasets, split.by = "stim")
Encontre características altamente variáveis para cada condição separadamente
# Identifique os 2.000 principais recursos variáveis para cada conjunto de dados na lista
sc_datasets.list <- lapply(X = sc_datasets.list, FUN = function(x) {
x <- FindVariableFeatures(x, selection.method = "vst", nfeatures = 2000) })
Encontre os recursos que são repetidamente variáveis em conjuntos de dados para integração (anchor features).
# Identificar recursos de integração a serem usados para alinhar conjuntos de dados
features <- SelectIntegrationFeatures(object.list = sc_datasets.list)
Encontre as âncoras de integração ou o conjunto final de genes altamente variáveis mais frequentemente selecionados nos lotes (batches)
# Encontre âncoras de integração entre os conjuntos de dados usando os recursos selecionados
sc_datasets.anchors <- FindIntegrationAnchors(object.list = sc_datasets.list, anchor.features = features)
# Abaixo está o resumo do processo em execução que aparecerá na sua tela
Integrar conjuntos de dados - Cria um ensaio de dados integrado
# Integre os conjuntos de dados em um único objeto Seurat usando as âncoras de integração
sc_datasets.combined <- IntegrateData(anchorset = sc_datasets.anchors)
# Abaixo está o resumo do processo em execução que aparecerá na sua tela
# Veja a estrutura do objeto Seurat integrado
dplyr::glimpse(sc_datasets.combined)
Execute o fluxo de trabalho padrão para visualização e agrupamento.
# Escalone os dados integrados, execute PCA, execute UMAP para redução de dimensionalidade, encontre vizinhos e agrupe células
sc_datasets.combined <- ScaleData(sc_datasets.combined, verbose = FALSE) %>%
RunPCA(npcs = 30, verbose = FALSE) %>%
RunUMAP(reduction = "pca", dims = 1:30) %>%
FindNeighbors(reduction = "pca", dims = 1:30) %>%
FindClusters(resolution = 0.5)
# Defina o tamanho dos gráficos para visualização
options(repr.plot.height = 5, repr.plot.width = 16)
# Visualização: Crie gráficos UMAP com diferentes opções de agrupamento ou rotulagem
p1 <- DimPlot(sc_datasets.combined, reduction = "umap", group.by = "stim") # Agrupe células por metadados "stim"
p2 <- DimPlot(sc_datasets.combined, reduction = "umap", label = TRUE, repel = TRUE) # Rotule clusters no UMAP
p3 <- DimPlot(sc_datasets.combined, reduction = "umap", group.by = "seurat_annotations") # Agrupe por anotações
p1 + p2 + p3 # Combine os gráficos