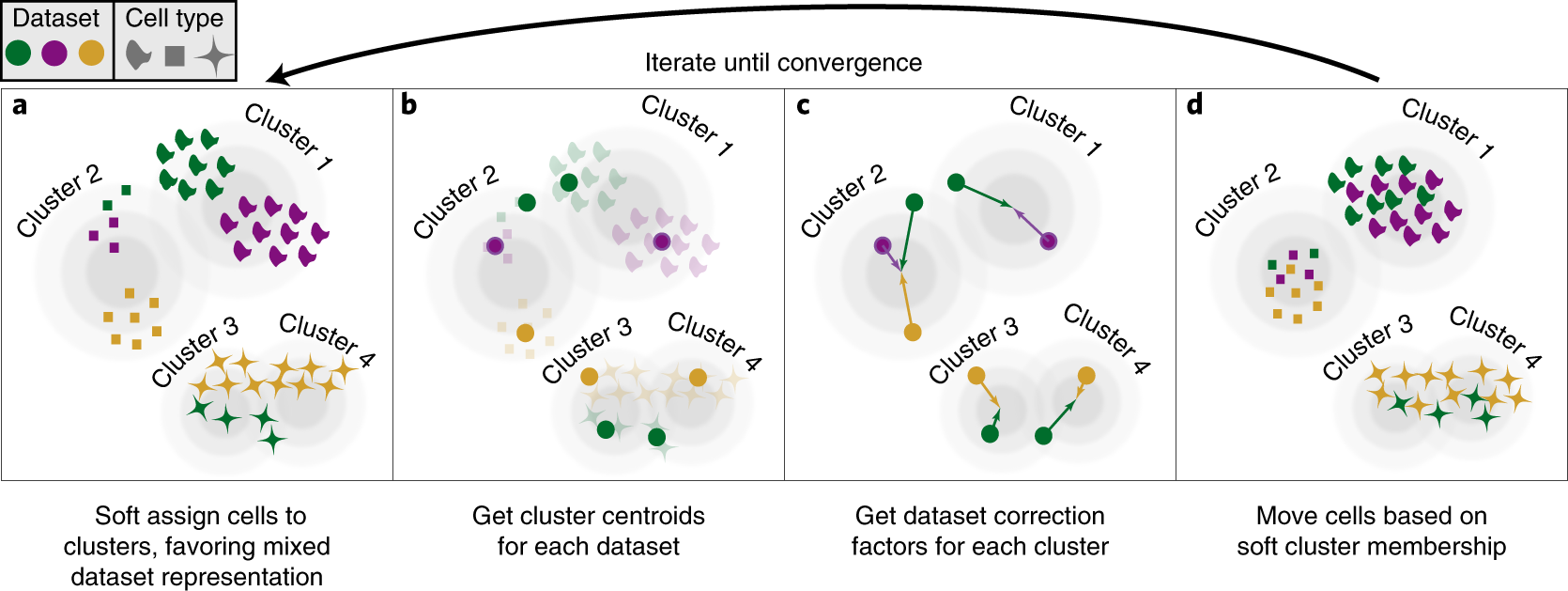
Run Seurat data integration
The integration performed here with Seurat consists of using a Canonical Correlation Analysis to identify anchors between datasets, as indicated in the original paper:
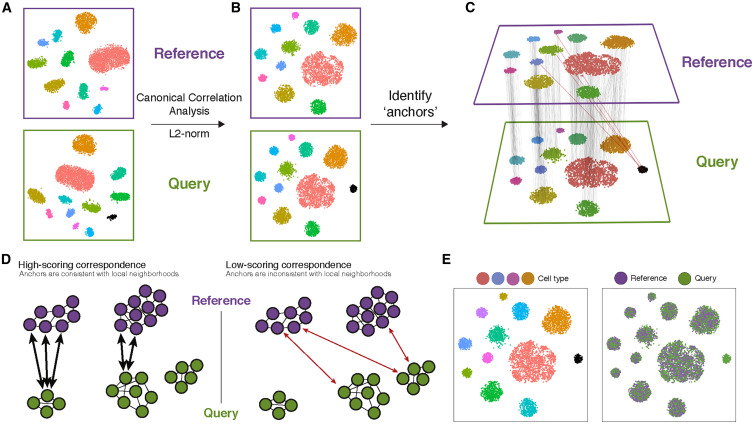
Split dataset into a list of datasets
# Identify the top 2000 variable features for each dataset in the list
sc_datasets.list <- lapply(X = sc_datasets.list, FUN = function(x) {
x <- FindVariableFeatures(x, selection.method = "vst", nfeatures = 2000)
})
Find highly variable features for each condition separately
# Identify integration features to be used for aligning datasets
features <- SelectIntegrationFeatures(object.list = sc_datasets.list)
Find the features that are repeatedly variable across datasets for integration (anchor features).
# Find integration anchors between the datasets using the selected features
sc_datasets.anchors <- FindIntegrationAnchors(object.list = sc_datasets.list, anchor.features = features)
# Below is the resume of the running process that will appear in your screen
Integrate datasets - Creates an integrated data assay
# Integrate the datasets into a single Seurat object using the integration anchors
sc_datasets.combined <- IntegrateData(anchorset = sc_datasets.anchors)
# Below is the resume of the running process that will appear in your screen
# View the structure of the integrated Seurat object
dplyr::glimpse(sc_datasets.combined)
Run the standard workflow for visualization and clustering.
# Scale the integrated data, run PCA, perform UMAP for dimensionality reduction, find neighbors, and cluster cells
sc_datasets.combined <- ScaleData(sc_datasets.combined, verbose = FALSE) %>%
RunPCA(npcs = 30, verbose = FALSE) %>%
RunUMAP(reduction = "pca", dims = 1:30) %>%
FindNeighbors(reduction = "pca", dims = 1:30) %>%
FindClusters(resolution = 0.5)
# Set the size of the plots for visualization
options(repr.plot.height = 5, repr.plot.width = 16)
# Visualization: Create UMAP plots with different grouping or labeling options
p1 <- DimPlot(sc_datasets.combined, reduction = "umap", group.by = "stim") # Group cells by "stim" metadata
p2 <- DimPlot(sc_datasets.combined, reduction = "umap", label = TRUE, repel = TRUE) # Label clusters on the UMAP
p3 <- DimPlot(sc_datasets.combined, reduction = "umap", group.by = "seurat_annotations") # Group by annotations
p1 + p2 + p3 # Combine the plots