Ejecutar la integración de datos con Seurat
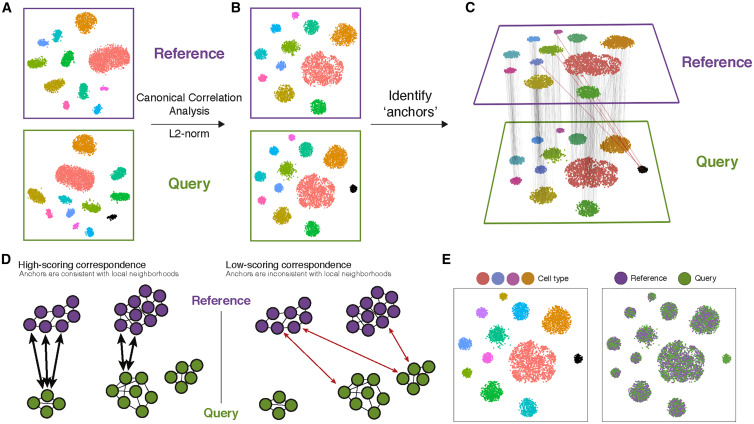
La integración realizada aquí con Seurat consiste en utilizar un Análisis de Correlación Canónica (CCA) para identificar anclas entre los conjuntos de datos, tal como se indica en el original paper:
Dividir el conjunto de datos en una lista de conjuntos de datos
# Identificar las 2000 características variables principales para cada conjunto de datos en la lista
sc_datasets.list <- lapply(X = sc_datasets.list, FUN = function(x) {
x <- FindVariableFeatures(x, selection.method = "vst", nfeatures = 2000)
})
Encontrar las características que son repetidamente variables entre los conjuntos de datos para la integración (características ancla).
# Identificar las características de integración que se usarán para alinear los conjuntos de datos
features <- SelectIntegrationFeatures(object.list = sc_datasets.list)
Encontrar las anclas de integración o el conjunto final de genes altamente variables seleccionados con mayor frecuencia entre los lotes.
# Encontrar anclas de integración entre los conjuntos de datos usando las características seleccionadas
sc_datasets.anchors <- FindIntegrationAnchors(object.list = sc_datasets.list, anchor.features = features)
# A continuación, se muestra un resumen del proceso en ejecución que aparecerá en tu pantalla
Integrar conjuntos de datos - Crea un ensayo de datos integrado
# Integrar los conjuntos de datos en un único objeto Seurat usando las anclas de integración
sc_datasets.combined <- IntegrateData(anchorset = sc_datasets.anchors)
# A continuación, se muestra un resumen del proceso en ejecución que aparecerá en tu pantalla
# Ver la estructura del objeto Seurat integrado
dplyr::glimpse(sc_datasets.combined)
Ejecutar el flujo de trabajo estándar para visualización y agrupamiento.
# Escalar los datos integrados, ejecutar PCA, realizar UMAP para reducción de dimensionalidad, encontrar vecinos y agrupar las células
sc_datasets.combined <- ScaleData(sc_datasets.combined, verbose = FALSE) %>%
RunPCA(npcs = 30, verbose = FALSE) %>%
RunUMAP(reduction = "pca", dims = 1:30) %>%
FindNeighbors(reduction = "pca", dims = 1:30) %>%
FindClusters(resolution = 0.5)
# Establecer el tamaño de los gráficos para la visualización
options(repr.plot.height = 5, repr.plot.width = 16)
# Visualización: Crear gráficos UMAP con diferentes opciones de agrupamiento o etiquetado
p1 <- DimPlot(sc_datasets.combined, reduction = "umap", group.by = "stim") # Agrupar células por metadatos "stim"
p2 <- DimPlot(sc_datasets.combined, reduction = "umap", label = TRUE, repel = TRUE) # Etiquetar los clústeres en el UMAP
p3 <- DimPlot(sc_datasets.combined, reduction = "umap", group.by = "seurat_annotations") # Agrupar por anotaciones
p1 + p2 + p3 # Combinar los gráficos