Instale a bibliotecas necessárias
# Baixe um script de shell para adicionar repositórios CRAN ao Ubuntu Jammy
download.file("https://github.com/eddelbuettel/r2u/raw/master/inst/scripts/add_cranapt_jammy.sh",
"add_cranapt_jammy.sh")
# Altere a permissão do arquivo para torná-lo executável
Sys.chmod("add_cranapt_jammy.sh", "0755")
# Execute the shell script
system("./add_cranapt_jammy.sh")
# Habilite o Binary Package Manager (bspm) para gerenciar pacotes do sistema e o CRAN
bspm::enable()
options(bspm.version.check=FALSE)
Vamos criar uma função R para realizar as chamadas do sistema
# Defina uma função para executar comandos shell e imprimir a saída
shell_call <- function(command, ...) {
result <- system(command, intern = TRUE, ...)
cat(paste0(result, collapse = "\n"))
}
Instale os pacotes necessários
# Instalar pacotes R necessários
install.packages("R.utils")
# Instalar Seurat Wrappers do GitHub (comentado)
# remotes::install_github('satijalab/seurat-wrappers@d28512f804d5fe05e6d68900ca9221020d52cf1d', upgrade=F)
# Instale o BiocManager se ainda não estiver instalado
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager", quiet = T)
# Instale o Harmony e o LIANA do GitHub
install.packages("harmony")
remotes::install_github('saezlab/liana', upgrade=F)
Introdução
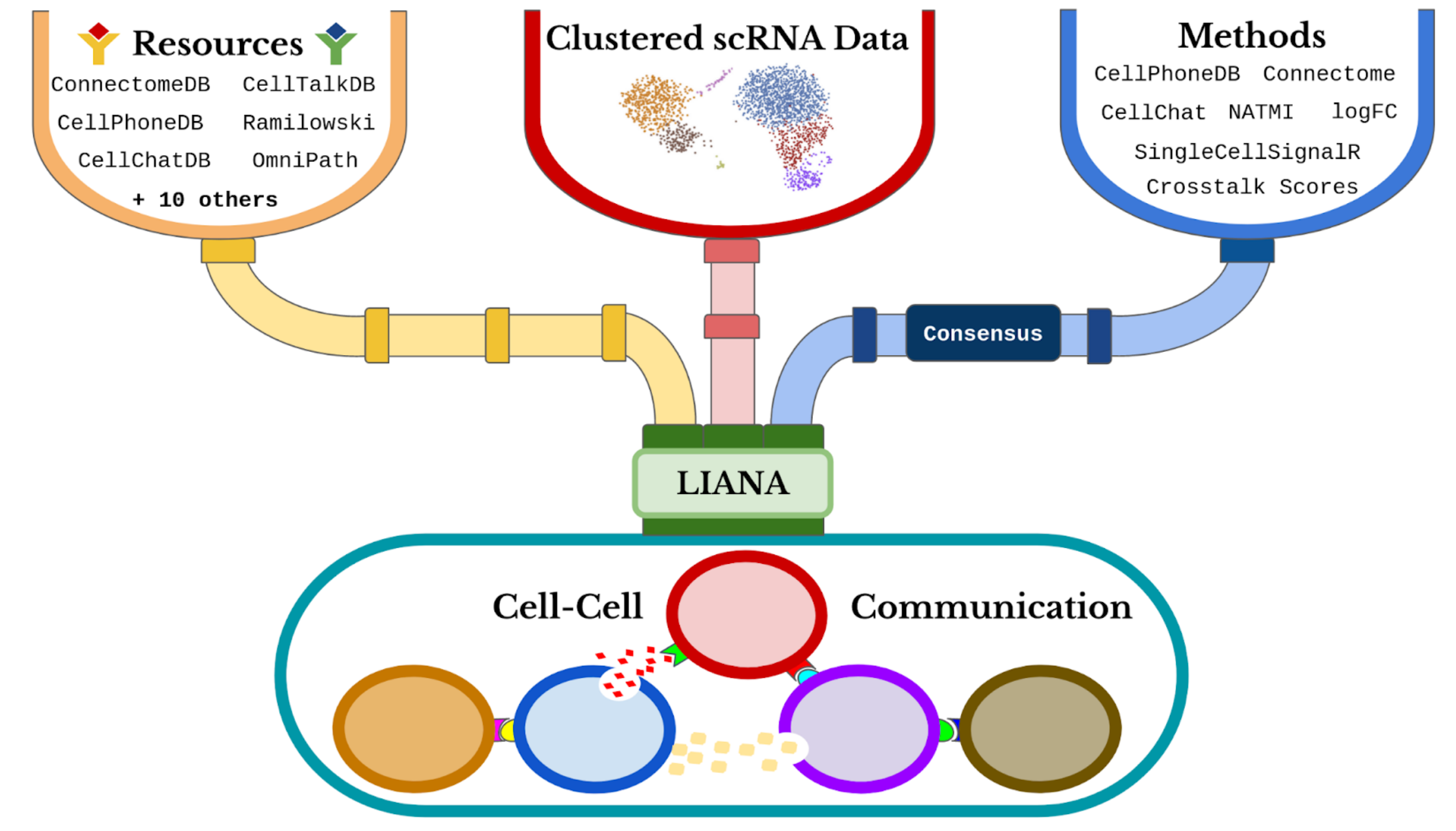
LIANA (Ligant-Receptor Inference Analysis) oferece uma variedade de métodos estatísticos para inferir interações entre ligante-receptor em dados transcriptômicos de células únicas a partir de conhecimentos prévios. Este notebook tem como objetivo demonstrar como usar o LIANA de forma básica com nossos dados de interesse.
Etapas para utilizar o LIANA:
# Instale o pacote Seurat para análise de células individuais
install.packages("Seurat")
# Carregar bibliotecas necessárias para manipulação e análise de dados
library(tidyverse) # Coleção de pacotes para ciência de dados
library(magrittr) # Operador de tubo
library(liana) # Análise da comunicação célula-célula
library(Seurat) # Análise de RNA-seq de célula única
Nesta seção, mostraremos todos os métodos implementados pelo LIANA a partir de várias ferramentas. Cada método infere interações ligante-receptor relevantes com base em diferentes suposições. Normalmente, cada método retorna duas pontuações para cada par ligante-receptor:
1.Pontuação de Magnitude (Força): Esta pontuação indica a força da interação.
2.Pontuação de Especificidade: Esta pontuação reflete o quão específica a interação é para um determinado par de identidades de células.
# Mostrar métodos disponíveis na sessão R atual
show_methods()
Os diferentes recursos de interações ligante-receptor podem ser encontrados aqui. O consenso integra todos os outros recursos.
# Mostrar recursos disponíveis na sessão R atual
show_resources()
Carregando os Dados
Aqui nós vamos carregar nossos dados de interesse para estudar a comunicação célula-célula
# Baixe o conjunto de dados COVID-19 (arquivo RDS) do Dropbox
download.file("https://www.dropbox.com/scl/fi/1ysew52kr8o2riahzubcw/BALF-COVID19-Liao_et_al-NatMed-2020.rds?rlkey=tg3tpn8la6oth25wvx3a22qt9&dl=1", "COVID.rds")
# Leia o arquivo RDS em uma variável chamada 'testdata'
testdata <- readRDS('COVID.rds')
# Exibe uma visão geral da estrutura 'testdata'
testdata %>% dplyr::glimpse()
# Esta condição específica 'group == "S" ' está sendo usada para filtrar as linhas. Somente as linhas em que o valor da coluna group for igual a "S" serão incluídas no subconjunto.
testdata <- subset(x = testdata, subset = group == "S")
# Esta função é usada para obter ou definir os identificadores de um objeto. A nova coluna é chamada "celltype"
Idents(testdata) <- "celltype"
# Normalize os dados de RNA-seq de célula única usando Seurat
testdata <- Seurat::NormalizeData(testdata, verbose = FALSE)
# Exibir a estrutura do conjunto de dados processado
testdata %>% dplyr::glimpse()
Rodando o LIANA
Para rodar o LIANA, você pode escolher qualquer um dos métodos disponíveis. Nesse exemplo, iremos utilizar a implementação do CellPhoneDB implementation.
A função liana_wrap é capaz de realizar múltiplos métodos, cada um operando com os recursos fornecidos. Se nenhum método for especificado, liana_wrap irá executar todos os métodos implementados em LIANA. Adicionalmente, o recurso consensus é utilizado como padrão.
cpdb_result <- liana_wrap(testdata, # Esta função do pacote LIANA é usada para realizar análises de interação célula-célula usando diferentes métodos
method = 'cellphonedb',
resource = c('CellPhoneDB'), # Especifica que o método CellPhoneDB será usado
permutation.params = list(nperms=100, # Define os parâmetros de permutação nperms=100: Número de permutações
parallelize=FALSE, # Indica se a execução deve ser paralelizada
workers=4), # Número de trabalhos a serem usados se a execução fosse paralelizada
expr_prop=0.05) # Proporção mínima de expressão para considerar uma interação como válida
# Exibir a estrutura dos resultados da interação do CellPhoneDB
dplyr::glimpse(cpdb_result)
Para rodar o LIANA usando múltidos métodos simultaneamente, você pode especificar o método desejado no argumento method parameter. Neste exemplo, nós utilizaremos CellPhoneDB, NATMI, SingleCellSignalR (sca) e a abordagem logFC.
# Execute uma análise de interação mais complexa usando vários métodos
complex_test <- liana_wrap(testdata,
method = c('cellphonedb', 'natmi', 'sca', 'logfc'),
resource = c('CellPhoneDB')) # Usar recurso CellPhoneDB
# Exibir a estrutura dos resultados da interação complexa
dplyr::glimpse(complex_test)
Uma das principais características do LIANA é que ele pode calcular uma classificação de consenso da previsão de todos os métodos empregados para analisar a comunicação célula-célula. Usando a função liana_aggregate() podemos usar todos os resultados de cada método usado na etapa anterior.
# Agregar resultados de interação em vários métodos
liana_consensus <- complex_test %>% liana_aggregate()
# Exibir a estrutura dos resultados de interação agregados
dplyr::glimpse(liana_consensus)
Gráficos de pontos podem ser gerados para interpretar facilmente pares importantes de ligante-receptor usados por pares de células remetente-receptor.
Aqui, pré-processamos os resultados do CellPhoneDB e, em seguida, os plotamos. Um filtro para usar apenas casos significativos é aplicado (valor P < 0,05).
cpdb_int <- cpdb_result %>%
# Selecione apenas interações com p-val <= 0,05
filter(pvalue <= 0.05) %>% # Isso reflete a `especificidade` das interações
rank_method(method_name = "cellphonedb", mode = "magnitude") %>% # Em seguida, classifique de acordo com `magnitude` (lr_mean neste caso)
distinct_at(c("ligand.complex", "receptor.complex")) %>% # Selecione as 20 principais interações (independentemente do tipo de célula)
head(20)
# Options(repr.plot.height = 12, repr.plot.width = 9)
# Plote as interações célula-célula usando a função dotplot do LIANA
scPlot <- cpdb_result %>%
inner_join(cpdb_int, # Selecione apenas as interações de interesse
by = c("ligand.complex", "receptor.complex")) %>% # Inverter o tamanho (baixo p-value/alta especificidade = maior tamanho dos pontos), adicionar um valor pequenos para evitar valores infinitos para os 0s.
mutate(pvalue = -log10(pvalue + 1e-10)) %>% # Transforme os valores de p em log10 negativo
liana_dotplot(source_groups = c("Epithelial"), # Crie um gráfico de pontos para os grupos de origem e destino especificados e defina o grupo de origem
target_groups = c("Macrophages", "NK", "B", "T", "Neutrophil"), # Especifique os grupos-alvo
specificity = "pvalue", # Use o valor p para especificar o tamanho dos pontos
magnitude = "lr.mean", # Use a razão logarítmica média para a cor dos pontos
show_complex = TRUE,
size.label = "-log10(p-value)") + theme(axis.text.x = element_text(angle = 90))
scPlot
# Salve o gráfico como uma imagem
ggsave("01-liana_dotplot.png", plot = scPlot, bg = "white", dpi = 600, width = 16, height = 9)
Da mesma forma, podemos explorar os resultados consensuais
# Options(repr.plot.height = 12, repr.plot.width = 9)
# Plote as 20 principais interações dos resultados agregados do LIANA
scPlot <- liana_consensus %>%
liana_dotplot(source_groups = c("Macrophages"),
target_groups = c("Macrophages", "NK", "B", "T", "Neutrophil"),
ntop = 20) + theme(axis.text.x = element_text(angle = 90))
scPlot
# Salve o gráfico
ggsave("02-liana_dotplot.png", plot = scPlot, bg = "white", dpi = 600, width = 16, height = 9)
No geral,o potencial das células para se comunicar pode ser computado. Aqui, podemos contar o número de interações significativas/importantes. Então, elas podem ser visualizadas por meio de um mapa de calor.
Da mesma forma, podemos comparar os resultados do CellPhoneDB vs consenso.
# Filtre interações com valor p ≤ 0,05 e plotar mapa de calor
liana_trunc <- cpdb_result %>% filter(pvalue <= 0.05)
# Plote e salve o gráfico
# png("03-heat_freq.png", bg = "white")
heat_freq(liana_trunc)
# dev.off()
# Filtre as interações consenso com classificação agregada ≤ 0,01 e plote mapa de calor
liana_trunc <- liana_consensus %>% filter(aggregate_rank <= 0.01)
# Plote e salve o gráfico
# png("04-heat_freq.png", bg = "white")
heat_freq(liana_trunc)
# dev.off()