Install the required libraries
# Download a shell script to add CRAN repositories to Ubuntu Jammy
download.file("https://github.com/eddelbuettel/r2u/raw/master/inst/scripts/add_cranapt_jammy.sh",
"add_cranapt_jammy.sh")
# Change the file permission to make it executable
Sys.chmod("add_cranapt_jammy.sh", "0755")
# Execute the shell script
system("./add_cranapt_jammy.sh")
# Enable Binary Package Manager (bspm) to manage system and CRAN packages
bspm::enable()
options(bspm.version.check=FALSE)
We will create an R function to performs system calls
# Define a function to execute shell commands and print the output
shell_call <- function(command, ...) {
result <- system(command, intern = TRUE, ...)
cat(paste0(result, collapse = "\n"))
}
Install required libraries
# Install required R packages
install.packages("R.utils")
# Install Seurat Wrappers from GitHub (commented out)
# remotes::install_github('satijalab/seurat-wrappers@d28512f804d5fe05e6d68900ca9221020d52cf1d', upgrade=F)
# Install BiocManager if not already installed
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager", quiet = T)
# Install Harmony and LIANA from GitHub
install.packages("harmony")
remotes::install_github('saezlab/liana', upgrade=F)
Introduction
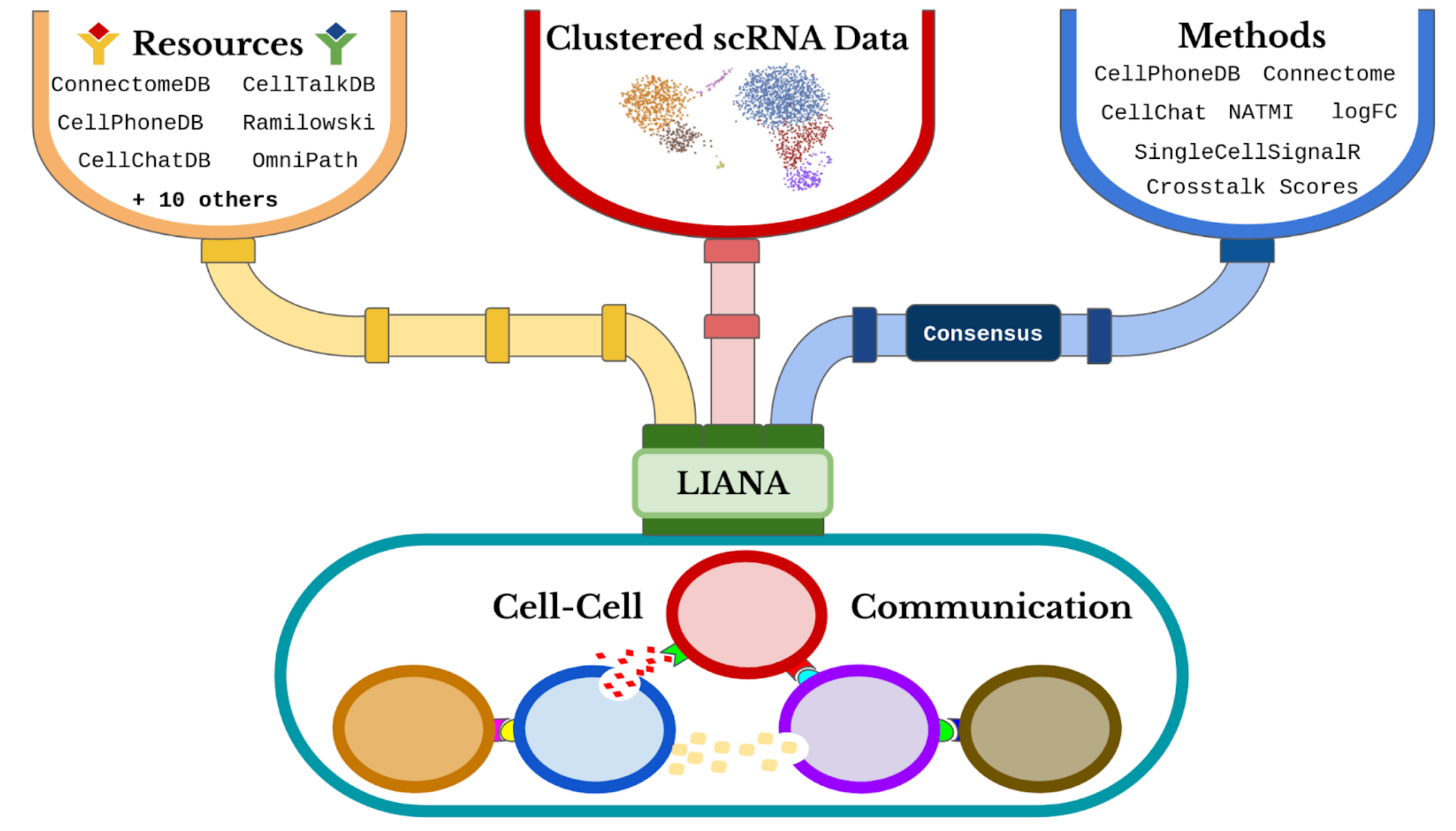
LIANA (Ligand-Receptor Inference Analysis) offers a variety of statistical methods to infer ligand-receptor interactions from single-cell transcriptomic data, using prior knowledge. This notebook aims to demonstrate how to use LIANA in a basic way with our data of interest.
Steps for Using LIANA:
# Install the Seurat package for single-cell analysis
install.packages("Seurat")
# Load necessary libraries for data manipulation and analysis
library(tidyverse) # Collection of packages for data science
library(magrittr) # Pipe operator
library(liana) # Cell-cell communication analysis
library(Seurat) # Single-cell RNA-seq analysis
In this section, we will showcase all the methods implemented by LIANA from various tools. Each method infers relevant ligand-receptor interactions based on different assumptions. Typically, each method returns two scores for each ligand-receptor pair:
1.Magnitude (Strength) Score: This score indicates the strength of the interaction.
2.Specificity Score: This score reflects how specific the interaction is to a given pair of cell identities.
# Show available methods in the current R session
show_methods()
The different resources of ligand-receptor interactions can be found here. The consensus integrates all the other resources.
# Show available resources in the current R session
show_resources()
Loading data
Here we load our data of interest to study cell-cell communicaton.
# Download the COVID-19 dataset (RDS file) from Dropbox
download.file("https://www.dropbox.com/scl/fi/1ysew52kr8o2riahzubcw/BALF-COVID19-Liao_et_al-NatMed-2020.rds?rlkey=tg3tpn8la6oth25wvx3a22qt9&dl=1", "COVID.rds")
# Read the RDS file into a variable called 'testdata'
testdata <- readRDS('COVID.rds')
# Display an overview of the 'testdata' structure
testdata %>% dplyr::glimpse()
# This specific condition 'group == "S" ' is being used to filter the rows. Only those rows where the group column value is equal to "S" will be included in the subset.
testdata <- subset(x = testdata, subset = group == "S")
# This function is used to get or set the identifiers of an object. The new columm is named "celltype"
Idents(testdata) <- "celltype"
# Normalize the single-cell RNA-seq data using Seurat
testdata <- Seurat::NormalizeData(testdata, verbose = FALSE)
# Display the structure of the processed dataset
testdata %>% dplyr::glimpse()
Running LIANA
To run LIANA, you can choose from any of the methods it supports. In this example, we will use the CellPhoneDB implementation.
The liana_wrap function invokes multiple methods, each operating with the provided resource(s). If no specific method is specified, liana_wrap will execute all methods implemented in LIANA. Additionally, the consensus resource is used by default.
cpdb_result <- liana_wrap(testdata, # This function of the LIANA package is used to perform cell-cell interaction analyses using different methods
method = 'cellphonedb',
resource = c('CellPhoneDB'), # Specifies that the CellPhoneDB method will be used
permutation.params = list(nperms=100, # Defines the permutation parameters. nperms=100: Number of permutations
parallelize=FALSE, # Indicates whether execution should be parallelized
workers=4), # Number of workers to use if execution were parallelized
expr_prop=0.05) # Minimum proportion of expression to consider an interaction as valid
# Display the structure of the CellPhoneDB interaction results
dplyr::glimpse(cpdb_result)
To run LIANA using multiple methods simultaneously, you can specify the desired methods in the method parameter. In this example, we will use CellPhoneDB, NATMI, SingleCellSignalR (sca), and the logFC approach.
# Run a more complex interaction analysis using multiple methods
complex_test <- liana_wrap(testdata,
method = c('cellphonedb', 'natmi', 'sca', 'logfc'),
resource = c('CellPhoneDB')) # Use CellPhoneDB resource
# Display the structure of the complex interaction results
dplyr::glimpse(complex_test)
One of the key features of LIANA is that it can compute a consensus ranking of the prediction of all methods employed to analyze cell-cell communication. By using the function liana_aggregate() we can use all the results from every method used in the previous step.
# Aggregate interaction results across multiple methods
liana_consensus <- complex_test %>% liana_aggregate()
# Display the structure of the aggregated interaction results
dplyr::glimpse(liana_consensus)
Dotplots can be generated to easily interpret important ligand-receptor pairs used by sender-receiver cell pairs.
Here, we preprocess the results of CellPhoneDB, then plot them. A filter to use only significant cases is applied (P-value < 0.05).
cpdb_int <- cpdb_result %>%
# Only keep interactions with p-val <= 0.05
filter(pvalue <= 0.05) %>% # This reflects interactions `specificity`
rank_method(method_name = "cellphonedb", mode = "magnitude") %>% # Then rank according to `magnitude` (lr_mean in this case)
distinct_at(c("ligand.complex", "receptor.complex")) %>% # Keep top 20 interactions (regardless of cell type)
head(20)
# Options(repr.plot.height = 12, repr.plot.width = 9)
# Plot cell-cell interactions using LIANA's dotplot function
scPlot <- cpdb_result %>%
inner_join(cpdb_int, # Keep only the interactions of interest
by = c("ligand.complex", "receptor.complex")) %>% # Invert size (low p-value/high specificity = larger dot size), add a small value to avoid Infinity for 0s
mutate(pvalue = -log10(pvalue + 1e-10)) %>% # Transforms the p-value by taking the negative log10
liana_dotplot(source_groups = c("Epithelial"), # Creates a dot plot for the specified source and target groups, and specifies the source group
target_groups = c("Macrophages", "NK", "B", "T", "Neutrophil"), # Specifies the target groups.
specificity = "pvalue", # Uses the p-value for dot size specification.
magnitude = "lr.mean", # Uses the mean log ratio for the dot color
show_complex = TRUE,
size.label = "-log10(p-value)") + theme(axis.text.x = element_text(angle = 90))
scPlot
# Save the plot as an image file
ggsave("01-liana_dotplot.png", plot = scPlot, bg = "white", dpi = 600, width = 16, height = 9)
Similarly, we can explore the consensus results.
# Options(repr.plot.height = 12, repr.plot.width = 9)
# Plot top 20 interactions from aggregated LIANA results
scPlot <- liana_consensus %>%
liana_dotplot(source_groups = c("Macrophages"),
target_groups = c("Macrophages", "NK", "B", "T", "Neutrophil"),
ntop = 20) + theme(axis.text.x = element_text(angle = 90))
scPlot
# Save the plot
ggsave("02-liana_dotplot.png", plot = scPlot, bg = "white", dpi = 600, width = 16, height = 9)
Overall potential of cells to communicate can be computed. Here, we can count the number of significant/important interactions. Then, they can be visualized through a heatmap.
Similarly, we can compare the CellPhoneDB vs consensus results.
# Filter interactions with p-value ≤ 0.05 and plot frequency heatmap
liana_trunc <- cpdb_result %>% filter(pvalue <= 0.05)
# Generate and save heatmap
# png("03-heat_freq.png", bg = "white")
heat_freq(liana_trunc)
# dev.off()
# Filter consensus interactions with aggregate rank ≤ 0.01 and plot heatmap
liana_trunc <- liana_consensus %>% filter(aggregate_rank <= 0.01)
# Generate and save heatmap
# png("04-heat_freq.png", bg = "white")
heat_freq(liana_trunc)
# dev.off()