Processing raw scRNA-seq data
This notebook introduces essential command-line operations in Linux, covering fundamental commands that are broadly applicable across programming languages with minimal adaptations. These foundational skills will support efficient data management and analysis in computational biology. Additionally, we will explore the key steps in processing raw sequencing reads into count matrices using Cell Ranger, discussing its main outputs and role in single-cell transcriptomics. Processing scRNA-seq data is a crucial step in single-cell analysis. The chosen library preparation method determines whether RNA sequences are captured from transcript ends (e.g., 10X Genomics, Drop-seq) or full-length transcripts (e.g., Smart-seq), directly influencing downstream analysis and biological insights.
Install Utilities
SRAtoolkit
NOTICE: Shell comands
Google Colab/Jupyter Noetbook using for default python as programming language. It's permite to use another languages, like shell script.
- For this, in Google Colab we use "!" before the code.
- For the Jupyter Notebooks we use a magic cell with %%bash before to write our script.
This is a marker to Google Colab understand this is Shell's code. For your personal use, "!" is not necessary.
%%bash
# A hashtag is a comment, this parte of the code is not using. However serve to importante annotations.
# It is a good and common practice in code as a reminder and as a mechanism for reproduction.
# We recommend to always comment your codes and script
echo "Hello, world!"
Along this Jupyter Notebook you will see diferents command in shell. They will be explained, but here is a small set of the most common commands.
%%bash
# make a folder.
# In programming, folder is called directory. We will use the name directory from here on.
# The name folder could be anything (e.g. folder1, folder_1, etc), try always to name directories that you remember what data are you saving on it
mkdir folder
%%bash
#list of files and directories
# In general, directories has a / in ending (e.g. /Documents/Files/scRNAseq_data/, here we have three directories
ls
%%bash
# A command could be follow for arguments. Arguments specify your main code. There are could be - or -- or positional.
# It's a importante thing to know about a software that you want to use.
ls -l
%%bash
# A command could be follow for arguments. Arguments specify your main code. There are could be - or -- or positional.
# It's a importante thing to know about a software that you want to use.
ls -l
%%bash
# The command cd is used to move into a directory or out of a directory
# If you want to move to another directory, you can use cd .. to move back forward or only cd to move forward
cd
%%bash
# The command mv is used to move your file to a specific directory
# You only need to specify the path to the directory you want to move your file
mv your_arquive local_to_move/
%%bash
# Also, mv could change names of directories or files. Try:
mv folder/ directory/
%%bash
# The command cat shows the entire contents of your file
cat
%%bash
# Also, you can concatenate two or more files using this command
cat file_1 file_2 > file_3
%%bash
# The command head shows the first 10 lines of your file
# Like a head of the "cat"
head your_file
%%bash
# The command tail shows the last 10 lines of your file
# Like a tail of the "cat"
tail
%%bash
# The command wget is used to download files
# You can put a link next to the command to download data (e.g. an specific dataset of a database, here is an example of a RNA-seq sample from SRA)
wget https://trace.ncbi.nlm.nih.gov/Traces/sra?run=SRR24765940
Installation
To begin, you must download SRAtoolkit in order to use the fastq-dump tool, which allows you to download SRA files.
The SRA Toolkit is a set of tools developed by the National Center for Biotechnology Information (NCBI) to access, download, and manipulate high-throughput data stored in the Sequence Read Archive (SRA)
The fastq-dump is one of the tools in the SRA Toolkit. It is used to extract data in FASTQ or FASTA format from SRA accessions. FASTQ is a common format for storing DNA and RNA sequencing data, which includes the sequence reads and their qualities. fastq-dump allows users to convert SRA files to FASTQ files, making it easier to process and analyze these data in other bioinformatics tools.
%%bash
# The -q is for quit mode, to don't provide all the output of wget, just run silently.
# The --output-document is to specify the name of the file where the downloaded content should be saved.
wget -q --output-document sratoolkit.tar.gz https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/3.1.0/sratoolkit.3.1.0-ubuntu64.tar.gz
# .tar.gz is a file that has been first packaged with tar and then compressed with gzip.
# tar command is used to pack or unpack files on Unix/Linux systems. The name "tar" comes from "tape archive"
# -xzf: Indicates that tar should extract the contents of a gzip-compressed archive.
tar -xzf sratoolkit.tar.gz
%%bash
# list of archives
ls
Cell Ranger
What is Cell Ranger?
Cell Ranger is a software suite developed by 10x Genomics for analyzing single-cell RNA sequencing (scRNA-seq) data generated from their Chromium platform. It processes the raw sequencing data into meaningful insights, including gene expression matrices, cell clustering, and other downstream analyses.
Note:
Cell Ranger requires more than 12GB of RAM to run, it does not work with Colab.
Download:Update the link if it has expired and then the relevant genomic reference.
%%bash
# -O: name of output
wget -O cellranger-10.0.0.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-10.0.0.tar.gz?Expires=1768912964&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA&Signature=FMtot0YFitTbWXvsuYSTGPfxYIoqyGJwO1ejvl9r1HJP~NfxT34ffOIelVvXO-iHGLS62iMM3TUV1H7xsRTZ5tPVjH3pW-fEmVyiyKjxuG18vABH0bqXNiIj8QtmsUa-Cfnvotmk1vqk8cuSODH61jDxD-~hIDwW0Fwwtkru1GfZLLUKKxceVZuCQedlTULYQrxy1w4Lyx8E0duJvSjLWhhtw1ZJOOz~lsmdkiJXbkSDyILmMqafiqRxKWF1KGv6nt-bAscAXWgdAz8hA73mNcOVIOMbrHE863aK5qsTdKRiX99DNfIe8rV5VButQzHqvWWRUNNGAaSa6ZJXnJSteQ__"
%%bash
wget "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2024-A.tar.gz"
Alternatively, you can also download version 10.0.0 from a GitHub repository we created as a backup, but first you need to remove the "#" symbol to uncomment it, as it's not necessary to run it if you click the download link from the 10x website.
%%bash
#wget https://github.com/integrativebioinformatics/scNotebooks/blob/main/scNotebooks-Resources/cellranger-10.0.0.tar.gz
IMPORTANT:
If your current version of cellranger is different from the one used in this Jupyter Notebook, you will need to update it in the code directory or download the backup from GitHub.
> Example: cellranger-9.0.1 --> cellranger-10.0.0
Installation
To work with certain organisms not available in the resources provided by 10x, such as bacteria, plants, and viruses, it is necessary to create a reference for CellRanger:
- cellranger-10.0.0/bin/cellranger is the path to the software
- The mkref command is used to create custom reference packages
- --genome: Creates a directory for the output files
- --fasta: Specifies the reference genome
- /genome/genome.fa: Is the path to the genome in .fa format
- --genes: Specifies the reference GTF file
- /gtf/genome.annotation.gtf: Path to the annotation file
Here's a small example of how to generate a reference, in this case for chromosome 22, from the 10x reference human genome.
First, we install samtools to manipulate the FASTA genome file and select chromosome 22.
%%bash
#apt-get update
apt-get install samtools -y
The `samtools faidx` function in particular extracts chromosome 22, and then we redirect the output to a file called GRCh38.CHR22.fa.
%%bash
samtools faidx refdata-gex-GRCh38-2024-A/fasta/genome.fa chr22 > GRCh38.CHR22.fa
%%bash
gunzip -c refdata-gex-GRCh38-2024-A/genes/genes.gtf.gz | grep "chr22" | gzip > CHR22.genes.gtf.gz
# gunzip is for descompress archives gzip
# -c: indicates that the decompressed output should be sent to standard output (stdout) instead of creating a decompressed file on the filesystem.
# refdata-gex-GRCh38-2024-A/genes/genes.gtf: is the path to the compressed GTF file that contains the genomic annotations.
# grep "chr22": filters the lines that contain "chr22", extracting them
# gzip > CHR22.genes.gtf.gz: compresses the filtered lines and saves them to a new file called CHR22.genes.gtf.gz.
# | (pipe) is an operator that takes the output of one command and uses it as input to another command.
Finally, we have the necessary files to generate the new reference and we can run cellranger.
%%bash
cellranger-10.0.0/bin/cellranger mkref --genome=GRCh38-CHR22 --fasta=GRCh38.CHR22.fa --genes=CHR22.genes.gtf.gz
# cellranger-10.0.0/bin/cellranger: is the path to the Cell Ranger executable.
# --genome=GRCh38-CHR22: Name of the reference genome being created.
# --fasta=GRCh38.CHR22.fa: Path to the FASTA file containing the reference genome sequences.
# --genes=CHR22.genes.gtf.gz: Path to the GTF file containing the genomic annotations.
Now, we have our new reference available, which we can work with.
DO NOT RUN!
-
cellranger-10.0.0/bin/cellranger is a path to the software
- count is used to count messenger RNA (mRNA) molecules
- --id= is a directory name for outputs
- --fastqs path to files to analyse
- --sample files to analyse
- --transcriptome is the reference
%%bash
#DO NOT RUN!
#This \ break a line and continue the code in another line
cellranger-7.2.0/bin/cellranger count --id=run_count_SRR11537950 \
--fastqs=SRR11537950 --sample=SRR11537950 \
--transcriptome=refdata-gex-GRCh38-2020-A
#--localcores=2 --localmem=12
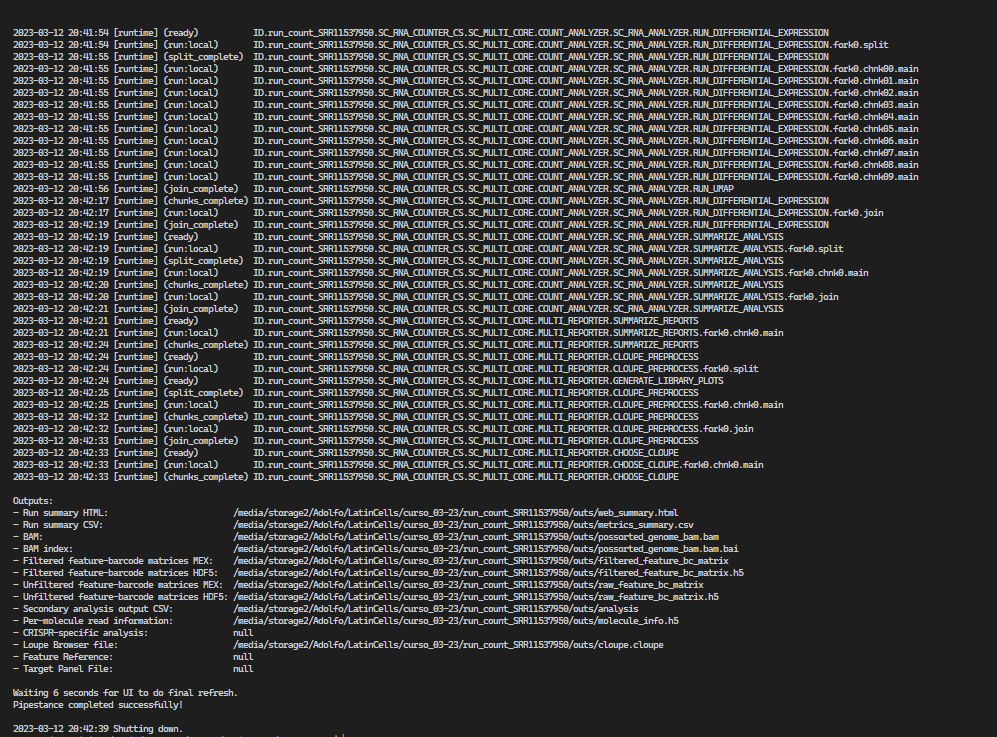
- The Cell Ranger software strives to maintain compatibility with common analysis tools by using standard output file formats whenever possible. For example, the barcoded BAM files can be viewed in standard genome browsers such as IGV to verify alignment quality and other features.
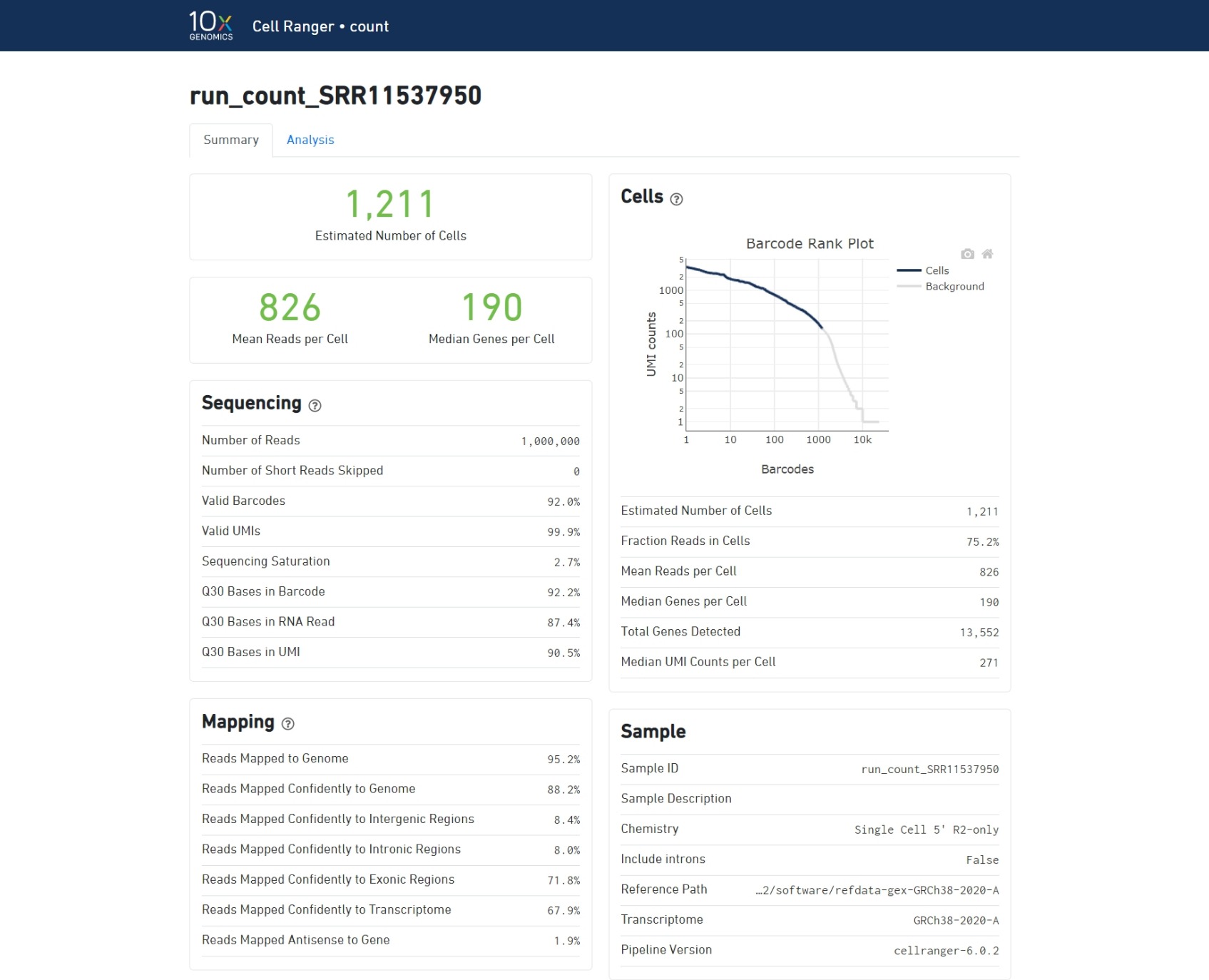
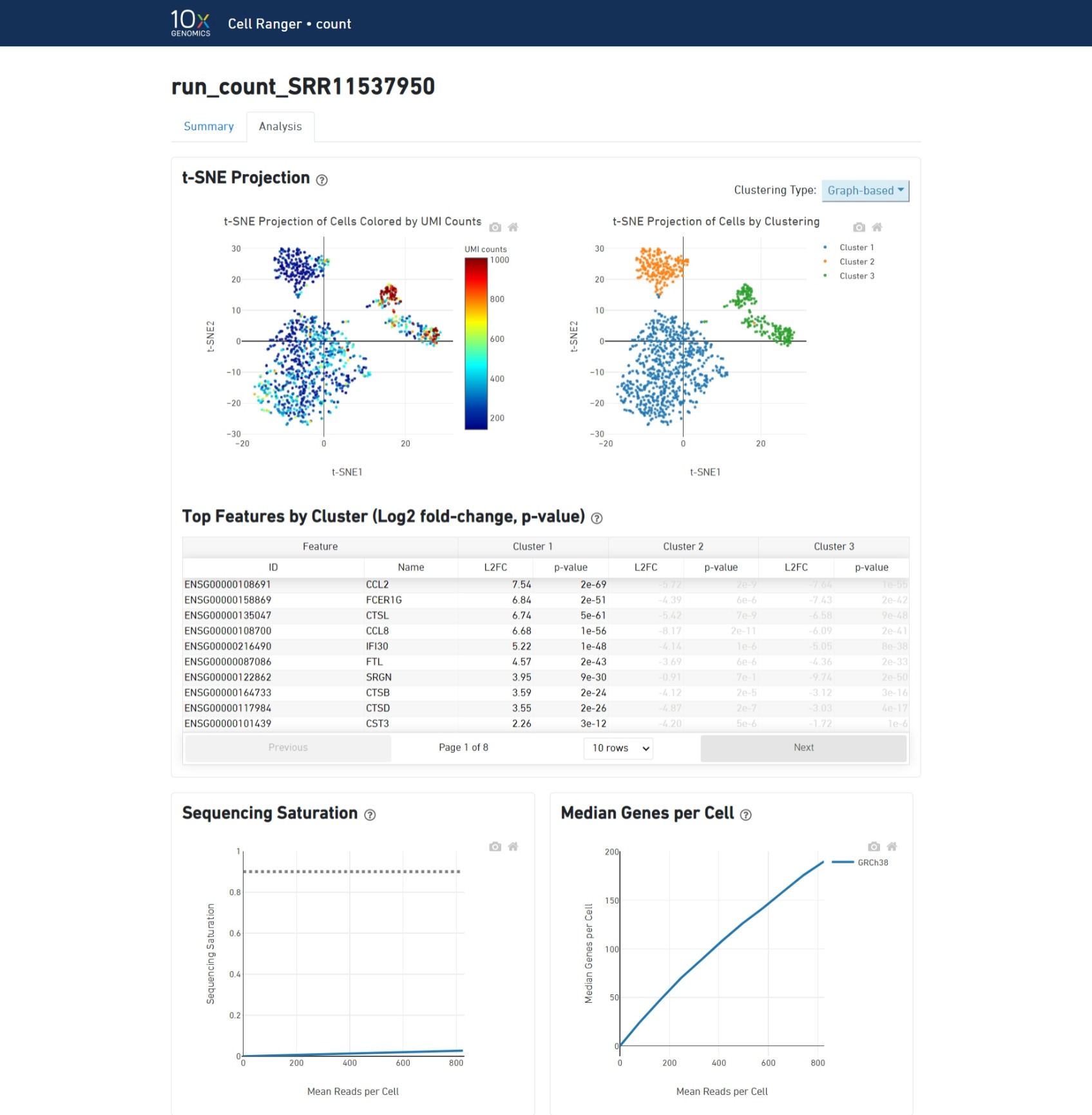
The cellranger count pipeline outputs an interactive summary HTML file named web_summary.html that contains summary metrics and automated secondary analysis results. Let's check it out!
https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/output/summary
- Green text indicates that the key metrics are in the expected range.
- Red/Yellow text indicates errors/warnings.
Understand the summary output:
Barcode Rank Plot:
A steep drop-off is indicative of good separation between the cell-associated barcodes and the barcodes associated with empty GEMs.